Overview
What is VAPER?
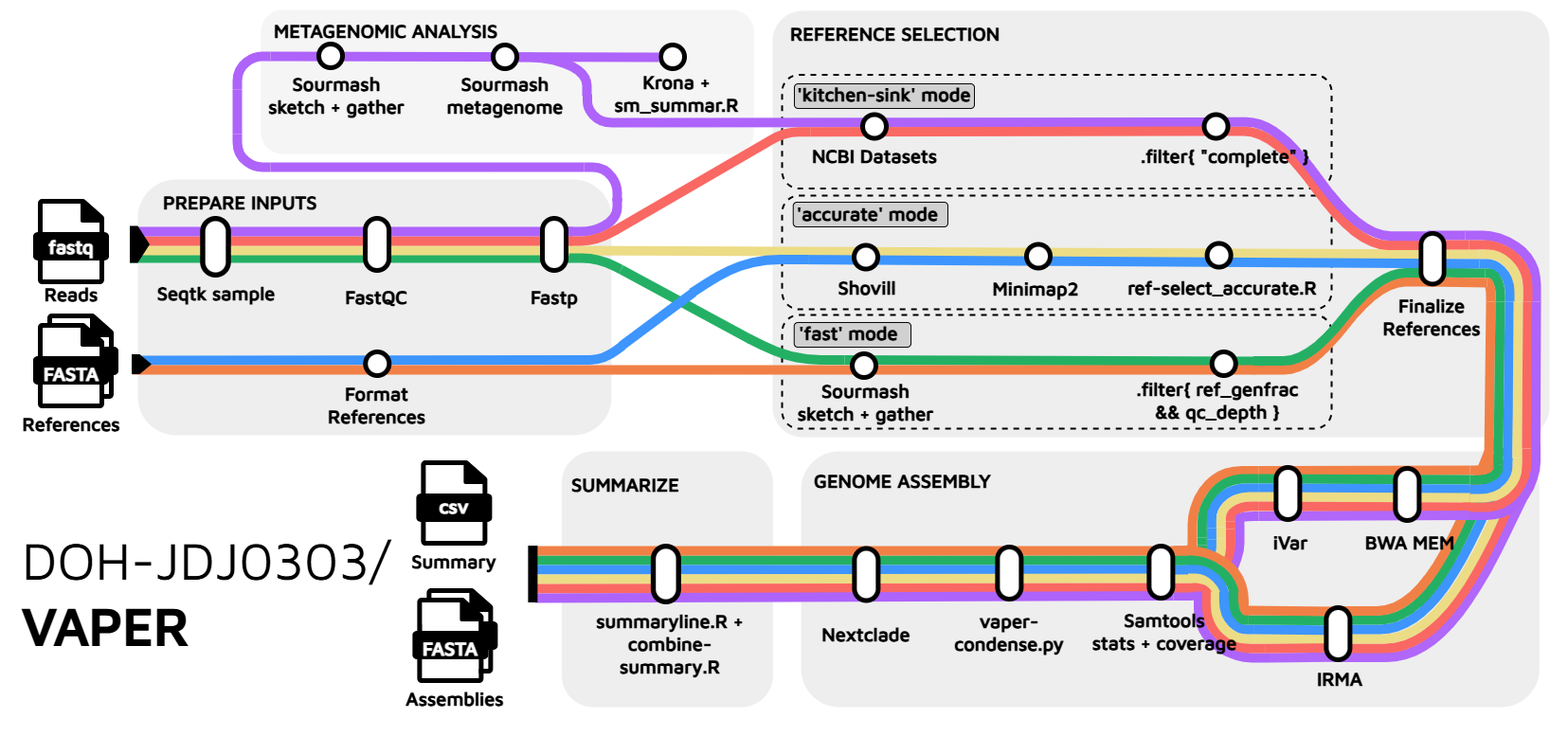
Flowchart
Key Features
VAPER (Viral Assembly from Probe-based Enrichment) is a viral (meta-)assembly pipeline that can:
🧬 Automatically detect and select reference genomes
🧬 Predict the taxonomy of each assembly, with an optional viral metagenomic summary
🧬 Export reads associated with each assembly for downstream use
While VAPER was originally designed for hybrid capture data (e.g., Illumina VSP or Twist CVRP), it has also been used with shotgun metagenomic and tile-amplicon data. It comes stock with comprehensive reference sets for [loading] taxa, including influenza A, SARS-CoV-2, and Monkeypox (full list). Keep on reading to learn more!
Contributors
VAPER was originally created by the Washington State Department of Health (WA DOH) as part of the Pathogen Genomics Center of Excellence (PGCoE). Check out the links below to learn more:
Inputs
VAPER performs basic quality control on the reads prior to reference selection and genome assembly. References are not quality controlled.
Read Quality
Read quality is evaluated and managed using FastQC and fastp. Fastp metrics are reported in the final summary. FastQC metrics can be found in the MultiQC report.
Read Downsampling
Reads can be optionally downsampled prior to read quality using the --max_reads parameter (default: 2000000 reads). This is accomplished using seqtk sample.
Downloading Reads from SRA
VAPER can download reads automatically from the NCBI Sequence Read Archive (SRA) (see example below).
samplesheet.csv
sample,sra
sample01,SRR28460430
These must be paired-end Illumina reads.
Reference Selection
VAPER can automatically select references for you and/or you can tell VAPER which references to use.
Automated Reference Selection
VAPER comes with multiple automated reference selection modes (--refs_mode ( accurate | fast | kitchen-sink )). accurate and fast modes use a reference set supplied using the --refs parameter. kitchen-sink mode downloads references from NCBI using the metagenomic summary.
Learn more about how reference sets are created here
accurate mode
As the name implies, reference selection using accurate mode is more accurate but slower. References are selected by mapping contigs from a de novo assembly to the entire reference set using minimap2 -x asm5 --secondary=no. The -x asm5 flag means that contigs will only map to references that share approx. 95% nucleotide identity. The --secondary=no flag means that contigs will only map to the closest matching reference (no multi-mapping). Together, these parameters allow VAPER to choose reference(s) that best match the sample. Tweaking the parameters below may improve results if you run into any issues:
--ref_genfrac: controls the minimum percent of a reference that must be mapped by one or more de novo contig for it to be selected for consensus generation.--denovo_assembler: controls which tool is used for de novo assembly. Options includemegahit,spades,velvet, andskesa.--denovo_contigcov: controls the minimum coverage required for a contig to be included in the de novo assembly.--denovo_contiglen: controls the minimum contig length required for a contig to be included in the de novo assembly.
fast mode
Also aptly named, fast mode is faster but less accurate (how much faster is up for debate). This approach uses sourmash gather to determine which reference(s) in the reference set are best represented in the raw reads. This mode has not been thoroughly tested. You can adjust this primarily using the --ref_genfrac parameter.
kitchen-sink mode 🚽
kitchen-sink mode attempts to build assemblies for each taxon identified in the metagenomic summary. Genome assemblies associated with each taxon are downloaded from NCBI using the NCBI datasets tool. Only assemblies that are listed as complete are included. Assemblies containing multiple contigs are split into individual FASTA files to avoid concatenating segmented viruses. As you can imagine, this method of reference selection can be very unreliable and should therefore be used with caution ⚠️.
Manual Reference Selection
You can tell VAPER which reference(s) to use for each sample by supplying a file path and/or the name of a reference in a reference set. This information is included in the references column of the samplesheet for each sample (see the examples below).
Example: File path
samplesheet.csv:
sample,fastq_1,fastq_2,reference
sample01,sample01_R1.fastq.gz,sample01_R2.fastq.gz,/path/to/ref.fa.gz
Example: Reference name
The example below would match reference Betacoronavirus-wg-1.fa.gz in the default reference set.
samplesheet.csv:
sample,fastq_1,fastq_2,reference
sample01,sample01_R1.fastq.gz,sample01_R2.fastq.gz,Betacoronavirus-wg-1
Example: Multiple references
Multiple references can be supplied for each sample using a semicolon (;). You can supply file paths and reference names at the same time 🙌!
samplesheet.csv:
sample,fastq_1,fastq_2,reference
sample01,sample01_R1.fastq.gz,sample01_R2.fastq.gz,Betacoronavirus-wg-1;/path/to/ref.fa.gz
Genome Assembly
VAPER creates genome assemblies by aligning reads to a reference genome and calling the consensus or plurality at each reference position (default: consensus). This can be accomplished using either ivar or irma (default: --cons_assembler ivar).
The de novo assembly used for ‘accurate’ reference selection is not used in the final reference-based assembly.
Assembly Parameters
Learn more about how to adjust assembly parameters here.
You can fine tune your IRMA assembly by adjusting the multitude of parameters located here: assets/IRMA_MODULE/init.sh. This file will be automatically copied into the work directory and used to build the IRMA module. However, MIN_AMBIG, MIN_CONS_SUPPORT, MIN_CONS_QUALITY, and SKIP_E must be adjusted from the command line using --cons_allele_ratio, --cons_allele_depth, --cons_allele_qual, and --cons_assembly_elong, respectively.
Assembly Quality
Assembly quality is evaluated using Nextclade and custom scripts. Quality metrics are reported relative to the reference genome used to create the assembly. Assemblies are automatically classified as PASS or FAIL based on the QC thresholds set using the --qc_depth and --qc_genfrac parameters. Assemblies will still be saved if they fail QC!
VAPER reports the number of bases in the sample missing from the termini of the reference in a separate column (ASSEMBLY_TERMINI_GAPS) because this is not included in the missing bases count by Nextclade.
Genome Assemblers
ivar assembler
iVar is the default assembler used by VAPER. Reads are aligned to the reference genome using BWA MEM and the alignment pileup is passed to ivar for consensus generation.
irma assembler
IRMA is a CDC-developed viral assembler with some nifty features 🌼. The main selling point with IRMA is that it can iteratively adjust the reference genome to more closely match the sample reads, therefore resulting in a more accurate assembly. This is accomplished using a liberal initial read alignment approach that allows for greater sample-reference divergence. IRMA can also elongate the reference during the refinement process, ideally resulting in a more “complete” assembly (--cons_elong true; use with caution ⚠️). Out of the box, IRMA is limited to a select number of species: influenza, ebolavirus, and coronavirus. This is because IRMA requires species-specific “modules” that contain a defined set of reference genomes. We can get around this by building these modules on the fly using the VAPER references 😎.
Another important difference with IRMA is that is produces multiple different assembly types, including a consensus assembly, like iVar, along with a plurality assembly and a padded assembly. Each have their own considerations (read more here). VAPER returns the consensus assembly by default (--cons_assembly_type consensus).
iVar and IRMA can perform differently depending on the input data. Learn more here.
Condensing Duplicate Assemblies
VAPER will occasionally produce multiple, near-identical assemblies (often >99.9% identity). This generally occurs when fragmented contigs from the de novo assembly map to multiple, closely related references (--ref_mode accurate only). VAPER uses a custom process (see vaper-condense.py) to identify and merge assemblies that are highly similar, helping reduce redundancy in the final output. This process uses MinHash-based comparisons (via Sourmash) to calculate pairwise distances between assemblies and groups them using hierarchical clustering. Assemblies within a user-defined similarity threshold (default 98% ANI; --cons_condist) are grouped, and the most complete and supported representative (based on coverage × depth) is selected from each cluster. The result is a condensed set of assemblies that preserves diversity while removing near-identical entries. This improves efficiency and interpretability in downstream analyses.
Metagenomic Classification
VAPER performs a basic viral metagenomic analysis using sourmash gather and sourmash tax metagenome with the 21-mer viral GenBank database. You can supply alternative database files using --sm_db and --sm_taxa.
- The static image and summary exclude unclassified sequences when calculating relative abundance. Sequences with relative abundance ≤ 1% are grouped into
Other. - The term metagenomic is used liberally here. A better term would probably be a metaviromic analysis, as it only includes viral species.
Example Outputs
Below are examples of the metagenomic output for a Twist Bioscience synthetic RNA control for Influenza A H1N1.
Krona Plot
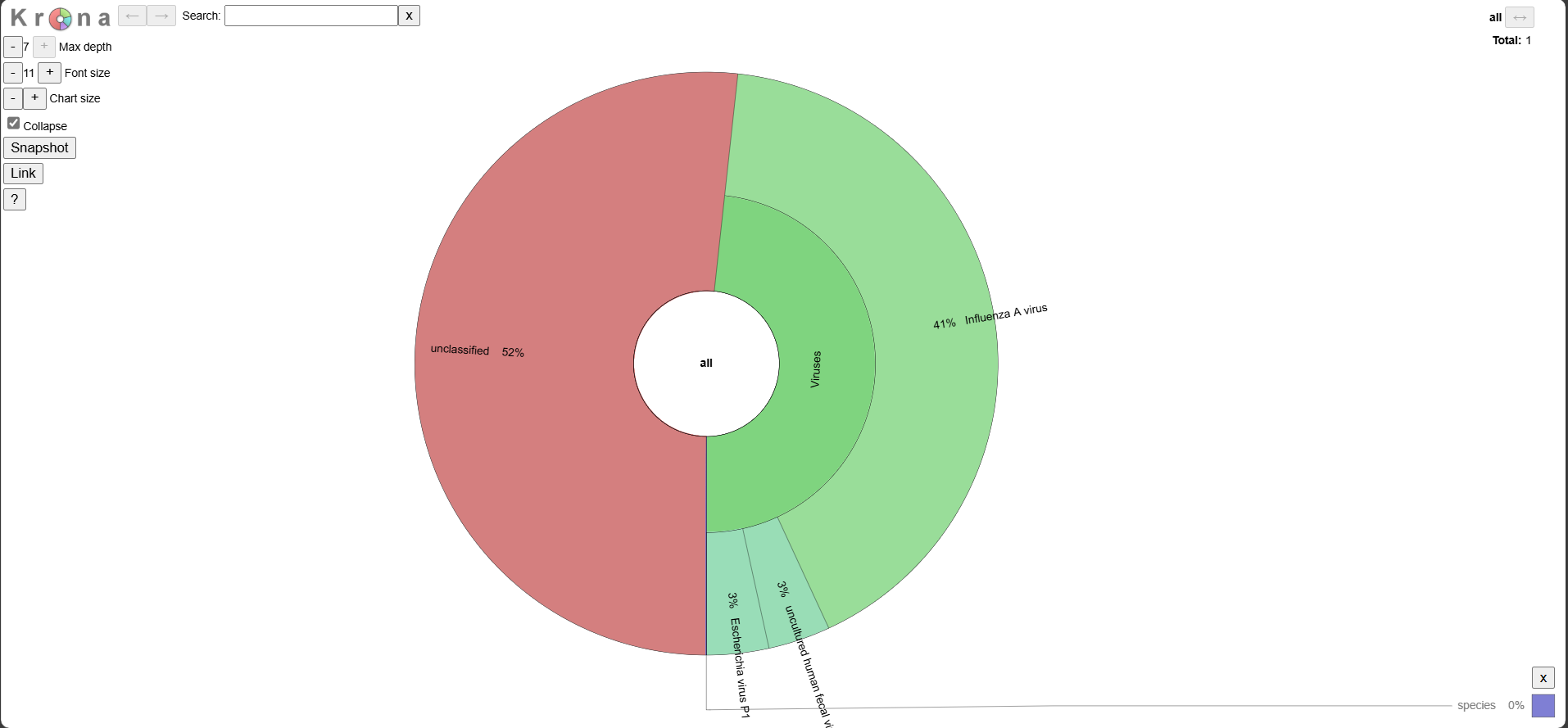 Explore the interactive plot here.
Explore the interactive plot here.
Static Image
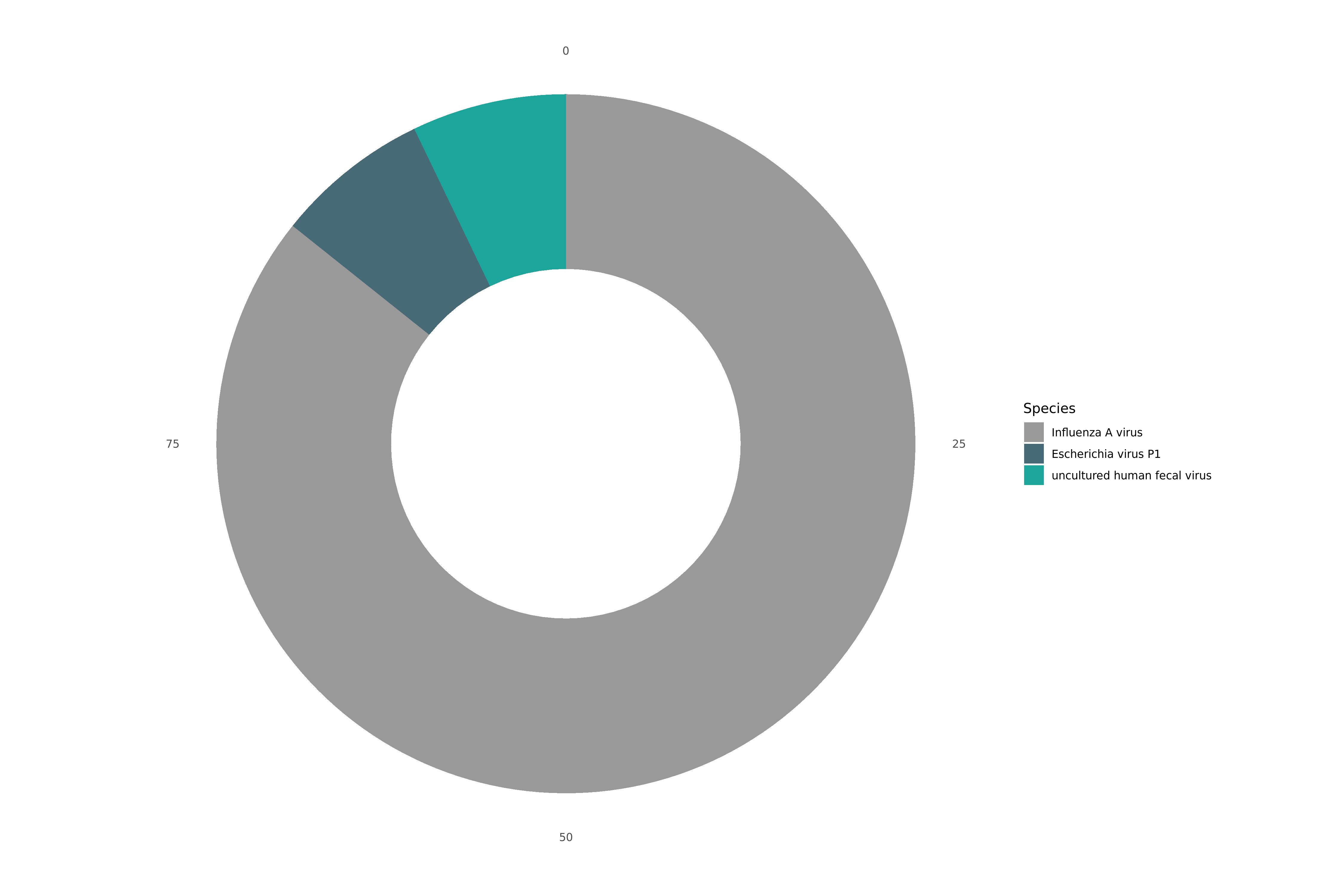
VAPER Summary
|SPECIES_SUMMARY| |:-| |91.2% Influenza A virus; 6.4% Escherichia virus P1; 6.4% uncultured human fecal virus|