Overview
What is VAPER?
Flowchart

Key Features
VAPER (Viral Assembly from Probe-based Enrichment) is a viral (meta-)assembly pipeline that can:
🧬 Automatically detect and select reference genomes
🧬 Predict the taxonomy of each assembly, with an optional viral metagenomic summary
🧬 Export reads associated with each assembly for downstream use
While VAPER was originally designed for hybrid capture data, it has also been used with shotgun metagenomic and tile-amplicon data. It comes stock with a comprehensive reference set for [loading] viral taxa, including all species targeted by the Illumina VSP v2.0 panel (see the full list here). Keep on reading to learn more!
Contributors
VAPER was originally created by the Washington State Department of Health (WA DOH) as part of the Pathogen Genomics Center of Excellence (PGCoE). Check out the links below to learn more:
Inputs
Read Downsampling
Reads can be optionally downsampled using the --max_reads parameter (default 2_000_000). This is accomplished using [seqtk][https://github.com/lh3/seqtk] and is primarily intended to control workflow efficiency. That said, this improved efficiency comes with a potential loss sensitivity, particularly during the reference selection stage. If you anticipate your target organism is present in a sample at low relative read depth, it is recommended that you use --ref_mode sensitive, which increases max_reads to 2 trillion reads (among other things).
Human Read Scrubbing
Human reads can be optionally removed using the --scrub_reads parameter (default false). This is accomplished using the SRA Human Read Scubber. Scrubbed reads can be found in each sample output directory: ${outdir}/${sample}/reads/
Read Quality
Read quality is evaluated and managed using FastQC and fastp. Fastp metrics are reported in the final summary. FastQC metrics can be found in the MultiQC report.
Downloading Reads from SRA
VAPER can download paired-end reads from the NCBI Sequence Read Archive (SRA) (see example below).
samplesheet.csv
sample,sra
sample01,SRR28460430
Reference Selection
VAPER can automatically select references for you and/or you can tell VAPER which references to use. Learn more about how to adjust reference selection parameters here. VAPER comes stock is a default reference set that includes thousands of viral species (see the full list here).
Automated Reference Selection
VAPER comes with multiple automated reference selection modes: --refs_mode ( standard | sensitive | kitchen-sink ). standard and sensitive modes use a reference set supplied using the --ref_set parameter. kitchen-sink mode downloads references from NCBI using the metagenomic summary.
Learn more about how reference sets are created here
Reference Selection Modes
Standard Mode
standardreference selection mode works as follows:
- A de novo assembly is created using Shovill
- Contigs are mapped to the reference set using Minimap2 (reference filters are applied at this stage - see next section)
- References covered by at least one contig are compared to each other with Sourmash and then clustered using DBSCAN and the nucleotide divergence threshold set by
ref_dist.- The reference with the greatest sample coverage is selected for each cluster and returned if its genome fraction meets the minimum set by
ref_genfrac.
parameter value max_reads 2_000_000 ref_genfrac 0.5 ref_denovo_contigcov 10 ref_denovo_contiglen 300 ref_denovo_depth 30
Sensitive Mode
sensitivereference selection uses the same method asstandardmode but with parameters adjusted to improve the detection of low abundance targets. This increased sensitivity is often met with a large decrease in efficiency (run times may increase dramatically! ⚠️).
parameter value max_reads 2_000_000_000_000 ref_genfrac 0.1 ref_denovo_contigcov 0 ref_denovo_contiglen 0 ref_denovo_depth 1000
Kitchen-Sink Mode 🚽
kitchen-sinkmode attempts to build assemblies for each taxon identified in the metagenomic summary. Genome assemblies associated with each taxon are downloaded from NCBI using the NCBIdatasetstool. Only assemblies that are listed as complete are included and those containing multiple contigs are split into individual FASTA files to avoid concatenating segmented viruses. As you can imagine, this method of reference selection can be very unreliable and should therefore be used with caution ⚠️.
accurate and fast reference selection modes used in VAPER v1.0 are now depricated. standard mode is like accurate mode but with more bells and whistles 🔔.
Reference Set Filtering
You can specify which references to select from using the ref_* filter columns / parameters. These filters can be set in the samplehseet (applied per sample) or the command line (applied to all samples). Below are examples of each method:
Sample-Level Filters
The example below shows how you would direct VAPER to only perform reference selection using Alphainfluenzavirus influenzae references for
sample01. This filter would not be applied tosample02. You could likewise filter by taxon, segment, and reference name using theref_taxon,ref_segment, andref_namecolumns.samplesheet.csv:sample,fastq_1,fastq_2,ref_species sample01,sample01_R1.fq.gz,sample01_R2.fq.gz,Alphainfluenzavirus influenzae sample02,sample02_R1.fq.gz,sample02_R2.fq.gz,
Run-Level Filters
The example below shows how you would direct VAPER to only perform reference selection using Alphainfluenzavirus influenzae references for all samples on the run. You could filter by taxon, segment, or reference name using the
--ref_taxon,--ref_segment, or--ref_nameparameters.nextflow run doh-jdj0303/vaper \ -r v2.0 \ -profile docker \ --input samplesheet.csv \ --outdir results \ --ref_species "Alphainfluenzavirus influenzae"
The ref_taxon, ref_species, and ref_segment filters control which references are considered during the selection process, whereas references specified by the ref_name filters will always be included, regardless of if there are reads to create an assembly.
Manual Reference Selection
References can also be supplied manually as individual file paths with or without an existing reference set. Like the reference set filters, this can be accomplished via the samplesheet (sample level) or the command line (run level). Multiple references can be supplied as a semicolon separated list. See below for examples:
Sample-Level Reference Files
samplesheet.csv:sample,fastq_1,fastq_2,ref_file sample01,sample01_R1.fq.gz,sample01_R2.fq.gz,/path/to/reference.fa.gz sample02,sample02_R1.fq.gz,sample02_R2.fq.gz,
Run-Level Reference Files
nextflow run doh-jdj0303/vaper \ -r v2.0 \ -profile docker \ --input samplesheet.csv \ --outdir results \ --ref_file /path/to/reference.fa.gz
Genome Assembly
VAPER creates genome assemblies by aligning reads to one or more reference and calling the consensus at each reference position. This is accomplished using BWA MEM, Samtools, and iVar.
VAPER v2.0 no longer supports the CDC IRMA assembler.
Learn more about how to adjust assembly parameters here.
Assembly Modes
VAPER comes with multiple default assembly modes. These modes primarily differ in how they handle reference positions with mixed read support.
The -t parameter used by iVar to control the minimum frequency threshold to make a consensus call does not behave as described in the documentation. Instead of controlling the frequency of bases needed to call a consensus, it actually controls the frequency of reads supported by the consensus, as described in this issue. Future versions of VAPER will work to address this limitation.
Standard Mode
standardmode returns the most common base observed among the reads (the plurality).
parameter value cons_allele_qual 20 cons_allele_ratio 0.0 cons_allele_depth 10 cons_max_depth 100
Mixed Mode
mixedmode includes minor allelic varation in the form of mixed IUPAC codes. This mode may be useful for environmental or pooled samples. The minimum and maximum read depth parameters are increased to account for multiple nucleotide calls at each position.
parameter value cons_allele_qual 20 cons_allele_ratio 0.8 cons_allele_depth 20 cons_max_depth 200
You can remove terminal Ns from each assembly using --cons_prune_termini. This is required when submitting some viral species to NCBI.
Assembly Quality
Assembly quality is evaluated using a custom script, vaper_stats.py. Quality metrics are reported relative to the reference genome used to create the assembly. Assemblies are automatically classified as PASS or FAIL based on the QC thresholds set using the --qc_depth and --qc_genfrac parameters.
Assemblies will still be saved if they fail QC!
Condensing Duplicate Assemblies
VAPER will occasionally produce multiple, near-identical assemblies (often >99.9% identity). This generally occurs when fragmented contigs from the de novo assembly map to multiple, closely related references. These assemblies are identified using a simple clustering approach (Sourmash + DBSCAN) and the minimum distance set by --cons_condist. Only the best assembly is returned, based on read coverage × depth.
Metagenomic Classification
VAPER performs an optional viral metagenomic analysis using sourmash gather and sourmash tax metagenome with the 21-mer viral NCBI database (Jan 2025). You can skip the metagenomic analysis using --metagenome false or supply alternative database files using --sm_db and --sm_taxa.
The static image and summary exclude unclassified sequences when calculating relative abundance. Sequences with relative abundance ≤ 1% are grouped into Other.
Example Outputs
Below are examples of the metagenomic output for a Twist Bioscience synthetic RNA control for Influenza A H1N1.
Krona Plot
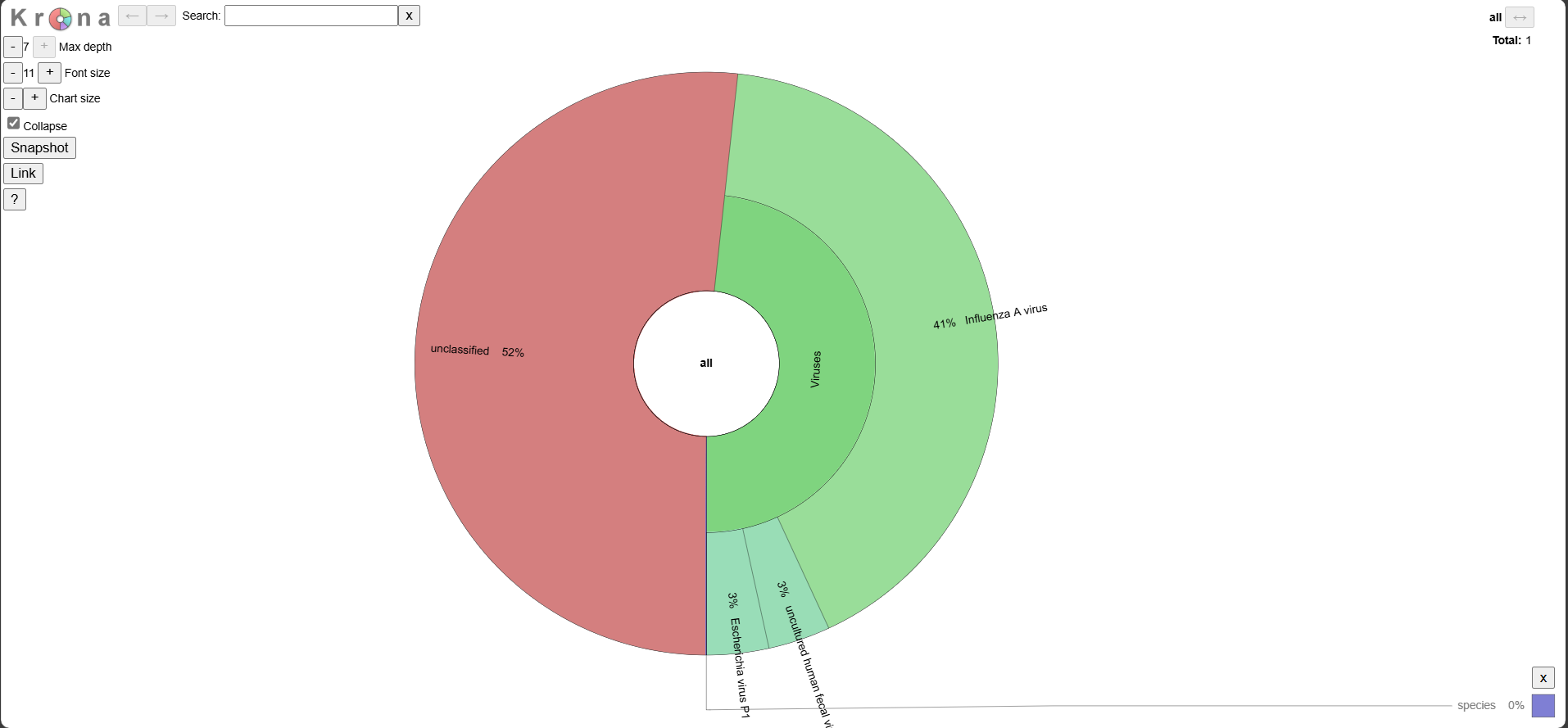 Explore the interactive plot here.
Explore the interactive plot here.
Static Image
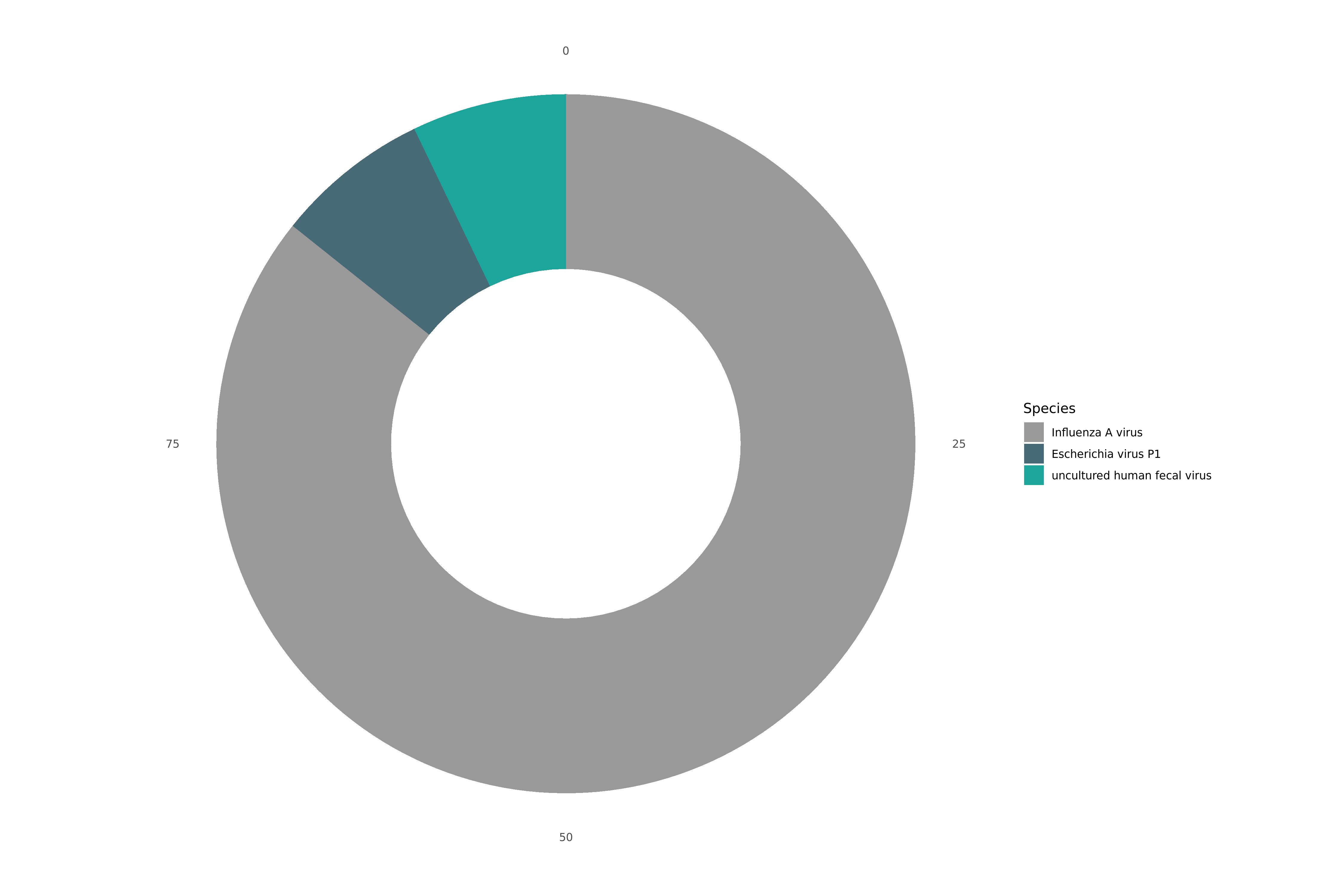
VAPER Summary
| SPECIES_SUMMARY |
|---|
| 91.2% Influenza A virus; 6.4% Escherichia virus P1; 6.4% uncultured human fecal virus |